
— Gezamenlijk onderzoek van Zhejiang CDC, Macro & Micro-Test en China CDC gepubliceerd in Frontiers in Cellular and Infection Microbiology
Studieoverzicht
In mei 2026 publiceerde Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4,6) een artikel onder leiding van het Zhejiang Provincial Center for Disease Control and Prevention (Zhejiang CDC), met het bio-informaticateam van Beijing Macro & Micro-Test Bio-Tech Co., Ltd. en het National Institute for Communicable Disease Control and Prevention (China CDC) als co-auteurs. De studie draagt de volgende titel:
“Identificatie en fylogenetische analyse van zeven Brucella abortus-stammen in Zhejiang, China.”

Deze studie is de eerste systematische, op het volledige genoom gebaseerde fylogenetische traceerbaarheidsanalyse van Brucella abortus (B. abortus) in de provincie Zhejiang, China. Het team analyseerde zeven isolaten die tussen 2015 en 2025 werden verzameld (vier stammen van menselijke oorsprong en drie van runderoorsprong uit Jinhua, Quzhou en Ningbo). De bevindingen leveren genomisch bewijs voor de oorsprong en transmissieroutes van deze "noordelijk dominante soort" in een atypisch zuidelijk epidemisch gebied in Oost-China.
Achtergrond en betekenis
Brucellose is een zoönotische ziekte die wordt veroorzaakt door bacteriën van het geslacht Brucella. Brucella abortus infecteert voornamelijk runderen, maar kan ook ziekte bij mensen veroorzaken. In China vertoont brucellose een duidelijke geografische variatie: de hoogste incidentie komt voor in de noordelijke provincies (bijvoorbeeld Binnen-Mongolië, Shanxi, Heilongjiang). Daarentegen wordt in de zuidelijke provincies, waaronder Zhejiang, historisch gezien gedomineerd door Brucella melitensis, met zeer weinig gerapporteerde gevallen van B. abortus. Deze regionale verschillen maken de genetische karakterisering en het traceren van de bron van B. abortus in Zhejiang een belangrijke prioriteit voor de volksgezondheid.
Methoden en belangrijkste bevindingen
Het onderzoeksteam hanteerde een veelzijdige strategie die moleculaire biologie en bio-informatica combineerde:
1.Identificatie van pathogenen en basistypering
PCR met het BCSP-31-gen en AMOS-PCR bevestigden dat alle zeven isolaten B. abortus waren.
Multilocussequentiebepaling (MLST) op basis van negen huishoudgenen toonde aan dat alle isolaten tot sequentietype ST2 behoorden, wat wijst op een hoge genetische homogeniteit onder de circulerende B. abortus-stammen in Zhejiang.
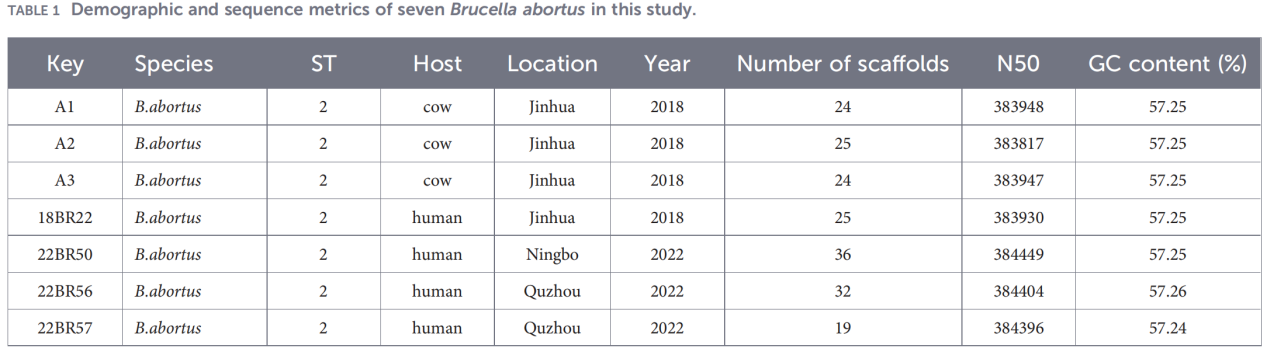
2.Karakterisering van het gehele genoom
Volledige genoomsequentiebepaling werd uitgevoerd op het Illumina NovaSeq-platform. Analyse van de gemiddelde nucleotide-identiteit (ANI) toonde aan dat de isolaten uit Zhejiang tot 99,99% overeenkomst vertoonden met de referentiestam B. abortus 544.
Pan-genoomanalyse bracht een zeer geconserveerde populatie aan het licht: er werden 3084 kerngenen geïdentificeerd, samen met slechts 10 schilgenen, en er werden geen zachte kern- of wolkgenen gedetecteerd.
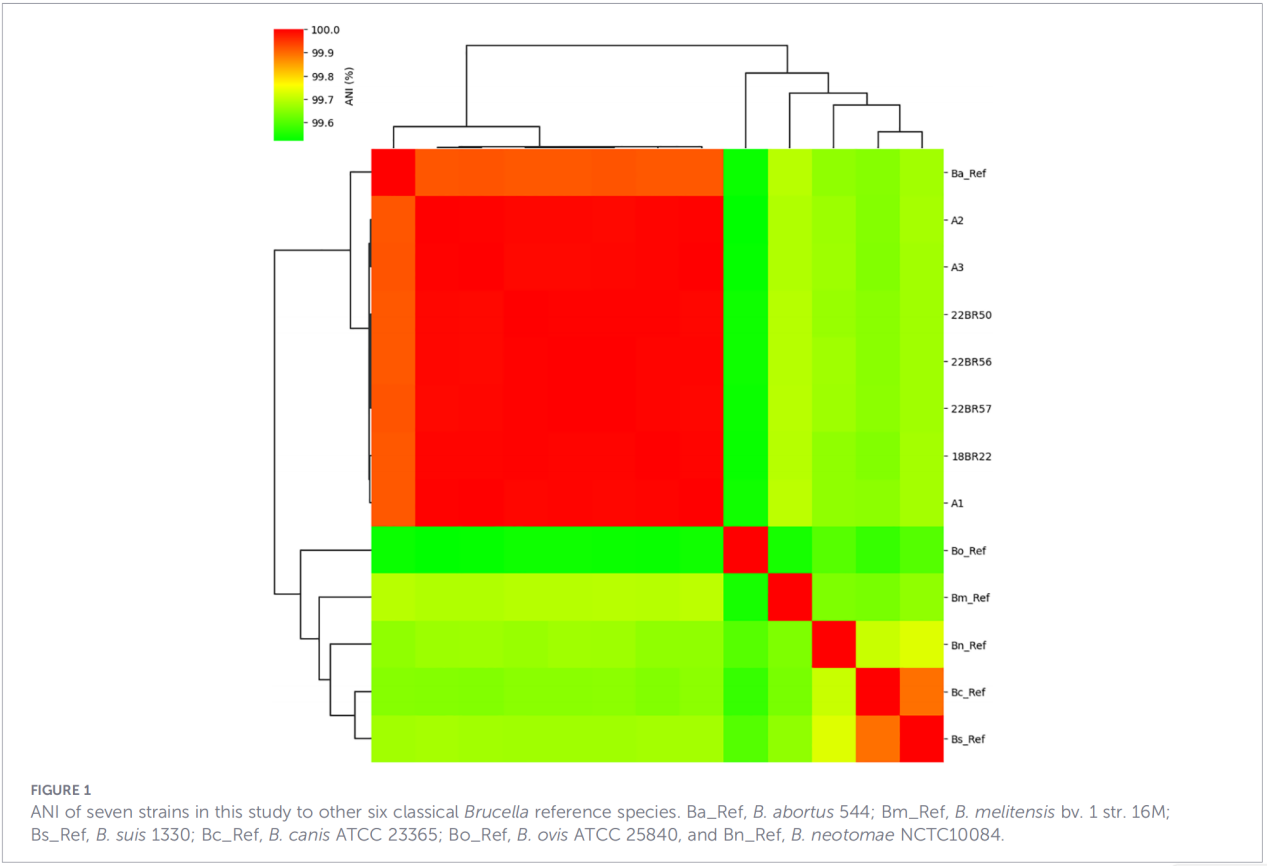
3.Genprofielen voor virulentie en antimicrobiële resistentie
In totaal werden 68 virulentiefactoren voorspeld, die klassieke pathways omvatten zoals LPS-biosynthese, het T4SS-secretiesysteem en het BvrR-BvrS-tweecomponentenregulatiesysteem. Opvallend is dat alle isolaten de adhesinegenen bmaA en btaF misten. Analyse van resistentiegenen detecteerde alleen het mprF-gen in de CARD-database; er werden geen andere resistentiebepalende factoren geïdentificeerd.
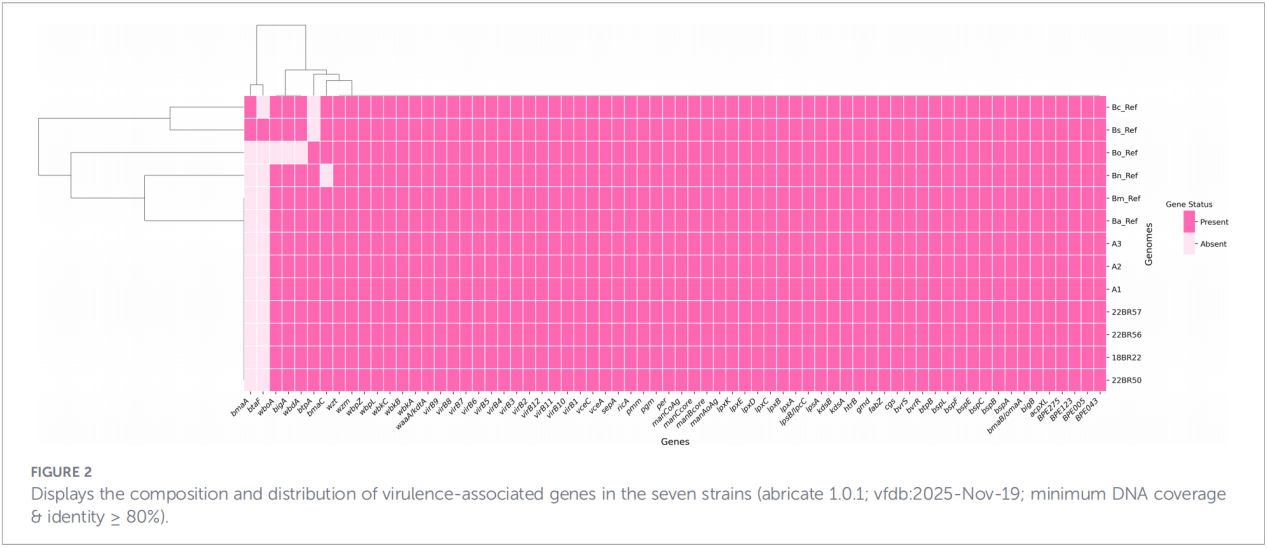 4. Fylogenetische reconstructie en transmissie-onderzoek
4. Fylogenetische reconstructie en transmissie-onderzoek
Analyse van single-nucleotide polymorfismen (cgSNP's) in het kerngenoom plaatste de isolaten uit Zhejiang op een specifieke positie in de wereldwijde fylogenetische boom. De resultaten toonden aan dat de stammen uit Zhejiang een monofyletische groep vormen samen met stammen uit Rusland, Mongolië en verschillende noordelijke Chinese provincies (Ningxia, Heilongjiang, Binnen-Mongolië, Hebei, Gansu, Beijing). Deze groep splitst zich verder op in drie afzonderlijke subgroepen (clade 1-3), wat wijst op meerdere onafhankelijke introductiegebeurtenissen.
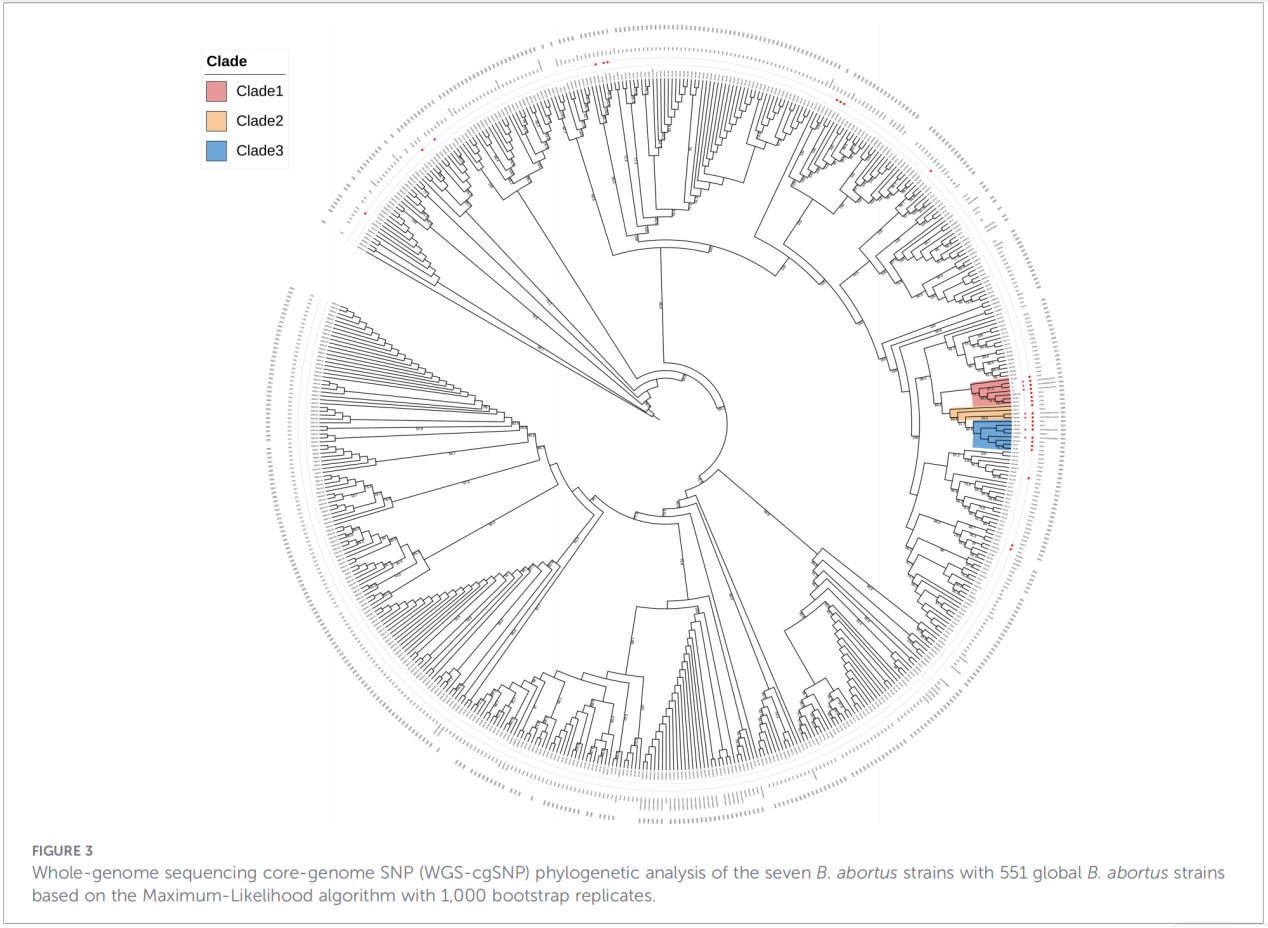
Conclusies en implicaties
Deze studie levert de eerste zeer nauwkeurige genomische dataset van B. abortus in de provincie Zhejiang op en leidt tot verschillende belangrijke conclusies:
- Clear genetische achtergrondDe B. abortus-stammen die in Zhejiang circuleren, behoren tot ST2, zijn genomisch sterk geconserveerd en vertegenwoordigen een typische runderbrucellose-lijn.
2. Evidichtheid van grensoverschrijdende transmissie– Fylogenetische analyse ondersteunt niet het bestaan van een onafhankelijke endemische lijn in Zhejiang. De gegevens suggereren daarentegen sterk dat deze stammen afkomstig zijn uit Noord-China en mogelijk een gemeenschappelijke evolutionaire achtergrond delen met stammen uit Rusland en Mongolië. De aanwezigheid van drie subgroepen duidt op meerdere afzonderlijke introductiegebeurtenissen.
3. Implicaties voor de volksgezondheid– De bevindingen onderstrepen de waarde van genomische surveillance voor brucellose, zelfs in traditioneel niet-endemische regio's zoals Zhejiang. Hoewel het huidige aantal gevallen laag is, kunnen zeer nauwkeurige methoden zoals cgSNP de bron van geïmporteerde uitbraken effectief traceren en wetenschappelijk bewijs leveren om transmissieketens te doorbreken die verband houden met interprovinciaal veetransport.
Dit onderzoek vult niet alleen een kennislacune in de provincie Zhejiang, maar levert ook nieuwe basisgegevens voor de surveillance van ziekteverwekkers en de risicobeoordeling van brucellose in de Yangtze-delta.
Papierinformatie:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identificatie en fylogenetische analyse van zeven Brucella abortus-stammen in Zhejiang, China. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Geplaatst op: 10 juni 2026